




宏基因组(Metagenomics),也称元基因组,利用新一代高通量测序技术(Next Generation Sequencingtechnology, NGS)以特定环境下微生物群体基因组为研究对象,在分析微生物多样性、种群结构、进化关系的基础上,可进一步探究微生物群体功能活性、相互协作关系及与环境之间的关系,发掘潜在的生物学意义。与传统微生物研究方法相比,宏基因组测序技术规避了绝大部分微生物不能培养、痕量菌无法检测的缺点,因此近年来在环境微生物学研究中得到了广泛应用。
通量高,拥有标准化实验室和高通量测序平台,数据库可靠
可检测不可培养物种,可检测痕量微生物
专业的生物信息团队,可以满足个性化的生物信息分析要求

▶ 样本要求
土壤:10 g/sample
粪便:3-5 g/sample
血液:10 mL/sample
污泥/沉积物:5-10 g/sample
DNA:总量≥1 μg,浓度≥100 ng/μL
▶ 检测平台
测序平台:Nova Seq 6000,Illumina
测序方法:PE150
测序深度:6G/10G/12G
▶ 常规项目周期
42个自然日
 医学领域:代谢病研究、肿瘤癌症研究等
医学领域:代谢病研究、肿瘤癌症研究等 畜牧领域:肠道、瘤胃(如产甲烷菌类群)与动物健康/营养消化研究等
畜牧领域:肠道、瘤胃(如产甲烷菌类群)与动物健康/营养消化研究等 农业领域:根际微生物与植物互作、农业耕作/施肥处理与土壤微生物群落研究等
农业领域:根际微生物与植物互作、农业耕作/施肥处理与土壤微生物群落研究等 环境领域:雾霾处理、污水治理、石油降解、酸性矿水处理及海洋环境研究等
环境领域:雾霾处理、污水治理、石油降解、酸性矿水处理及海洋环境研究等 生物能源:特殊功能的菌株、基因挖掘、工程菌的开发研究
生物能源:特殊功能的菌株、基因挖掘、工程菌的开发研究 特殊极端环境:极端环境条件下的微生物类群研究
特殊极端环境:极端环境条件下的微生物类群研究
▶ 宏基因组和代谢组分析揭示肠道菌群明显的结直肠癌阶段性特异表型
研究对象: 人
期刊: Nature Medicine
影响因子: 30.641
时间: 2019年
▶ 研究背景
大多数散发性结直肠癌的发生是多步过程,首先形成息肉样腺瘤,然后发展为粘膜内癌,最后发展为恶性肿瘤,其形成需要历经几十年的时间,早期癌症的发现和内镜切除是癌症控制有效手段,肠道菌群与结直肠癌的发生有密切关系。
▶ 研究结果
基于宏基因组和代谢组研究平台,对来自大队列CRC中的616名和406名样本分别进行了粪便宏基因组和代谢组学研究,获得不同阶段(MP, S0, SI /SII, III/SIV, HS)病例特异性表型的微生物(Atopob iumparvulum和B. wadsworthia等)和代谢(氨基酸和胆汁酸等)标志物;且进一步探索个体直肠癌患者的肠道微生物组与肿瘤与特征之间的关系。
▶ 结果展示
(1)宏基因组和代谢组分析
宏基因组分析首先发现Bacteroides和Prevotella是所有受试者和健康对照中变化最大的菌。进一步的研究还确定了与CRC关联的新物种,其中Colinsella aerofaciens, Dorealongicatena, Porphyromonasuenonis和Streptococcus anginosus等在III/IV中显著升高。代谢组分析发现,大肠中主要能源物质丙酸盐和丁酸盐是含量最高的两种代谢物,二氢尿嘧啶和尿素也存在较大差异。与健康对照组相比,MP中DCA (脱氧胆酸盐)浓度显著升高; S0中甘氨胆酸盐和牛磺胆酸盐浓度显著升高;支链氨基酸,苯丙氨酸,酪氨酸,甘氨酸,丝氨酸浓度也升高。
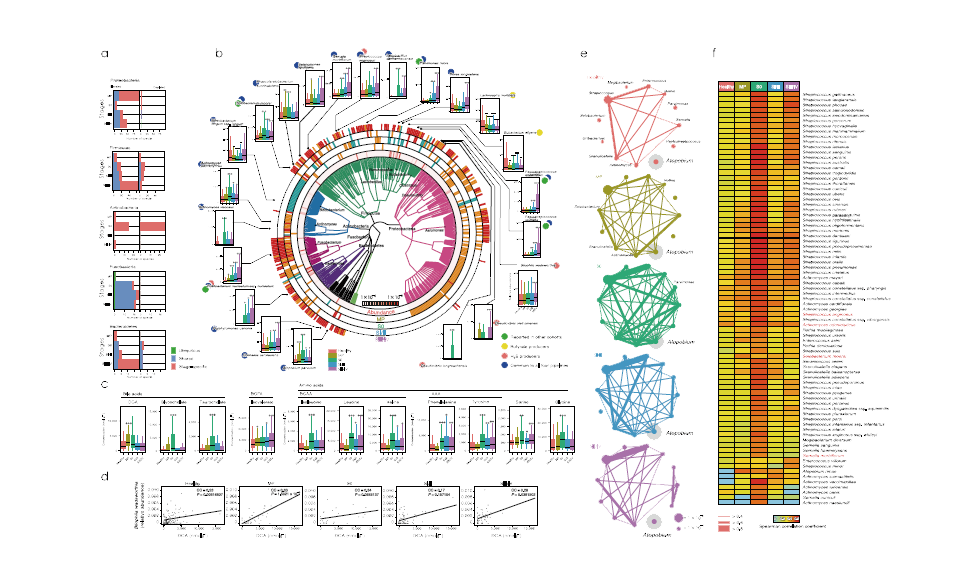
图1.肠道菌群与代谢物变化
(2)功能分析和标志物筛选
基于KEGG数据库筛选出1243个同源基因(KO基因)在至少一个阶段显著升高,96个KO基因在至少一个阶段显著降低。在差异最显著的通路中,芳香族氨基酸代谢和产硫化物的通路与CRC相关。参与苯丙氨酸和酪氨酸合成的基因显著升高,其中pheC被确定为区分S0患者和健康对照的得分最高标志物。通过构建随机森林和LASSO逻辑回归模型来区分健康对照与S0和SIII/IV。在S0分类中,特征值是包括编码环己二烯基脱水酶的pheC的KO基因;在SIII /IV分类中,特征值为已被确认为CRC标志物的P. micra,P. stomatis和 F. nucleatum等口腔厌氧菌。
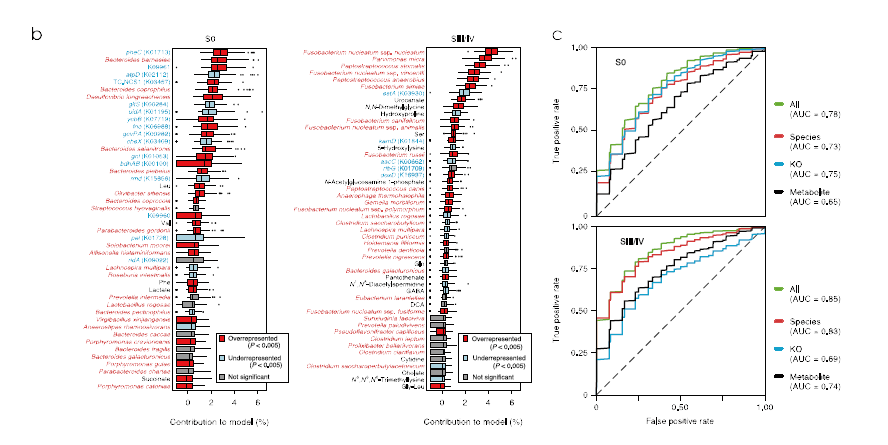
图2.标志物筛选
(3)结果验证和潜在机制
对28名CRC患者(SI / II和SIII / IV)术前和术后1年的粪便样品进行宏基因组测序。结果表明,肿瘤切除后P. stomatis, P. anaerobius和P. uenonis的相对丰度降低。
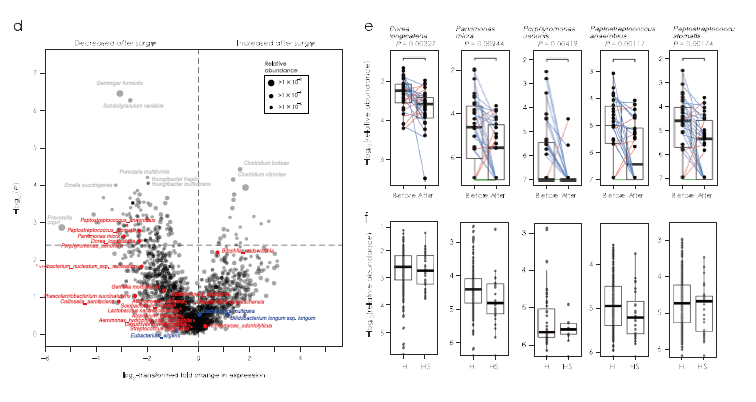
图3.与CRC相关微生物和代谢物验证结果
▶ 参考文献
Yachida S et al., Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med. 2019 doi: 10.1038/s41591-019-0458-7.
![]() 农学方向—案例分析
农学方向—案例分析
▶ 代谢组学和宏基因组学研究不同添加剂对青贮品质的影响
期刊: Bioresource Technology
影响因子: 9.642
时间: 2020年
▶ 研究背景
人口增长促进了全球肉类和牛奶消费量增加,养殖所需饲料用量也逐年上升,发酵后的饲料是进行农副产品处理更好的选择。罐头厂每年丢弃大量的破碎甜玉米,颗粒中含有大量的水溶性碳水化合物、淀粉和水分,但高含量的水分制约着青贮品质。一些青贮添加剂,如化学试剂、酶、乳酸菌等微生物可以促进发酵,提高营养价值,改善牲畜健康。虽然已有研究发现添加剂的使用影响青贮饲料中细菌群落的组成和相对丰度,但对青贮中的抗性基因和代谢物的影响研究较少。
▶ 研究结果
基于LC-MS非靶代谢组学和宏基因组技术平台,对四种不同处理的高水分玉米粒青贮(去离子水CK、香兰素V、植物乳杆菌LP和短乳杆菌LB,n = 6/组)进行研究,并发现不同添加剂对青贮的代谢物、微生物群落、病毒和抗生素抗性基因分布均有一定的影响。
(1)添加剂对微生物群落的影响
对甜玉米青贮饲料表面微生物和内部微生物都进行了宏基因组测序。发现添加剂显著影响青贮饲料中的微生物组成。甜玉米粒青贮的界、门、属和种水平的微生物群落如图所示。除了已知的细菌和真菌外,青贮饲料中还存在各种病毒和古菌。香兰素处理青贮样品中病毒的比例最高(2.7%),LP、LB和对照处理青贮样品中病毒的比例分别为1.3%、1.2%和1.5%。
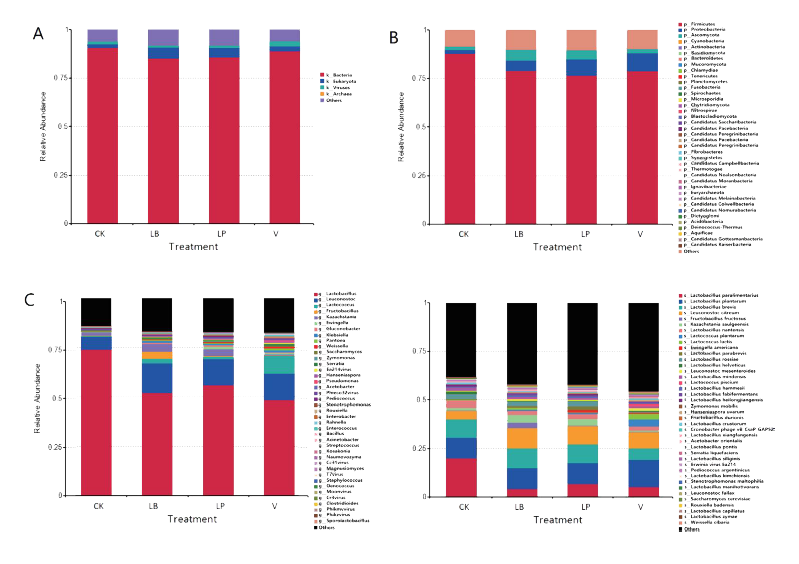
(2)添加剂对抗生素耐药性(ARO)的影响
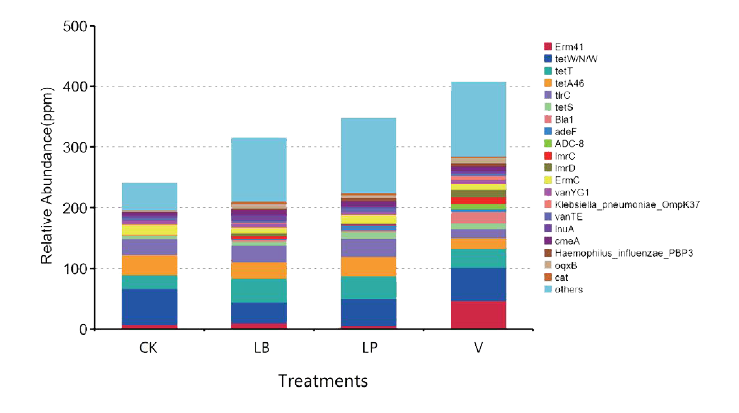
青贮饲料中添加的抗微生物化合物和微生物产生的次生代谢产物都对ARO的耐药菌组成和最终含量产生影响。虽然发酵特性表明V是一种很有前景的青贮添加剂,但它的应用可能会导致ARO在食用青贮饲料的动物肠道中传播。
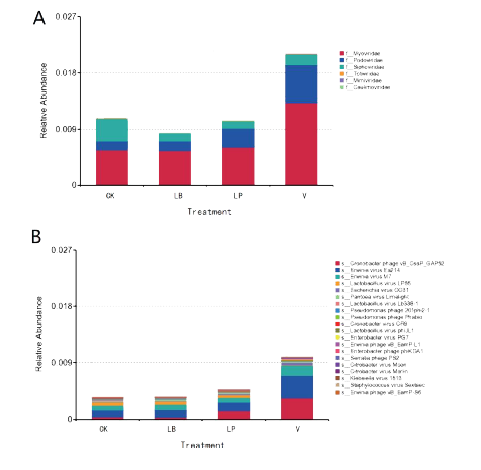
(3)添加剂对病毒的影响
本文是首次发现青贮中病毒的差异;对于病毒是如何影响青贮中的微生物群落的未进行深入研究。但初步确定病毒通过传播毒力因子和抗生素抗性基因提高毒力/抗性性状的表达。因此,高病毒含量可能是V处理青贮饲料中ARO基因丰度升高的另一个相关因素。
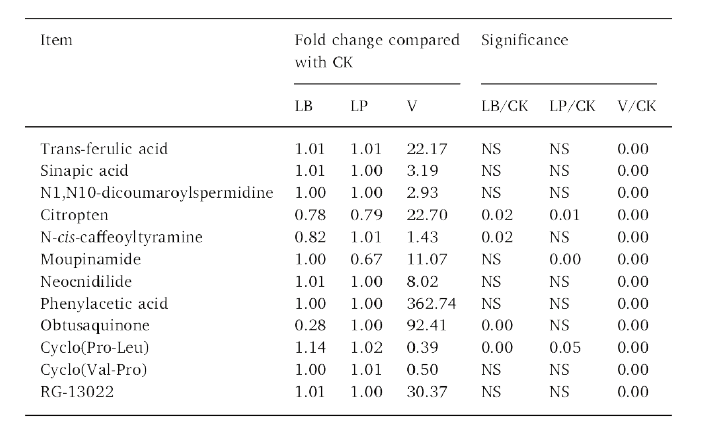
(4)添加剂对代谢物的影响
LB和LP处理青贮的代谢物的含量分别高于对照组青贮的含量。此外,与对照组青贮相比,V处理增加了93个代谢物的含量,降低了139个化合物的含量,而LP和LB处理分别降低了14和16个代谢物的含量。在青贮35天后,V处理的青贮样品比对照处理的青贮样品检测到更多的肉桂酸及其衍生物。
▶ 参考文献
Additives affect the distribution of metabolic profile, microbial communities and antibiotic resistance genes in high-moisture sweet corn kernel silage.








